
In all stages of drug discovery, machine learning models can be utilized to make faster and more insightful data-driven decisions. During model generation, all the available descriptors for a data set are typically generated irrespective of their importance (which may not be initially known). The resulting trained models are used to predict properties, which are intended to be useful in novel drug compound design or in compound optimization. A computational chemist or a cheminformatics expert will typically generate many models based on these full sets of descriptors, followed by the selection of a model that most accurately and robustly predicts the desirable compound property.
Challenges in model building
The industry does offer users with a variety of algorithms to select based on their needs and data type, yet many users struggle with most of them lacking a good user-interface (UI) that saves all the work being done programmatically. While some are struggling doing all of this at a code level others need a reliable management and versioning tool to control the production level models. Naming these models even manually is impossible in many applications.
Effortless model generation with Trainer Engine
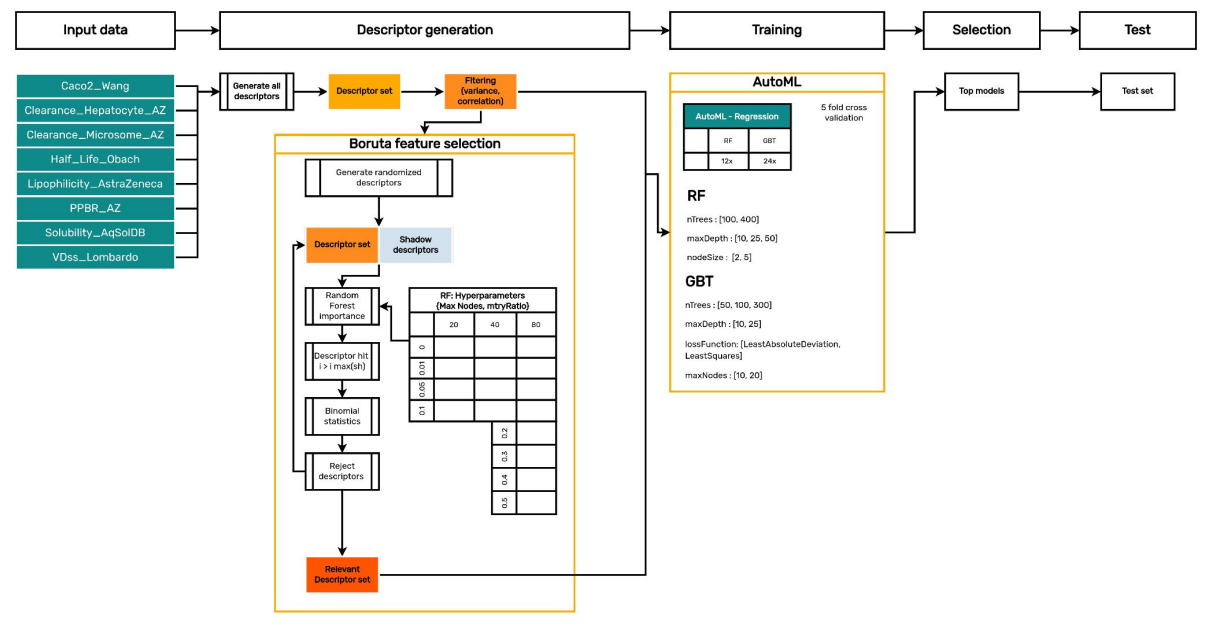
Chemaxon’s Trainer Engine can support the above-mentioned workflow, and also provides an interface which is user-friendly and easy to use. The “Automated Model building” (AutoML) workflow within Trainer Engine allows any researcher with limited or no experience to effortlessly generate multiple models (Random forest, Gradient boosted trees etc.). This model building capability only requires a few manual inputs and can be summarized by the following steps, where steps 2 through 5 are fully automated:
- User provides a small-molecule data set, an observed value to predict, and selects whether the method is regression or classification, and then begins the run.
- Compound structures in the dataset are checked and standardized.
- All available descriptors (~2500: a combination of proprietary Chemaxon and OpenSource descriptors) are generated.
- The Boruta Algorithm1 reduces the set of descriptors to the most relevant descriptor subset.
- The AutoML feature trains various models using different hyperparameters on the same data set.
- User analyzes the generated models through the analysis page and either refines the models’ parameters and re-runs them, or selects the best built model and deploys it to production.
Time and resource efficiency
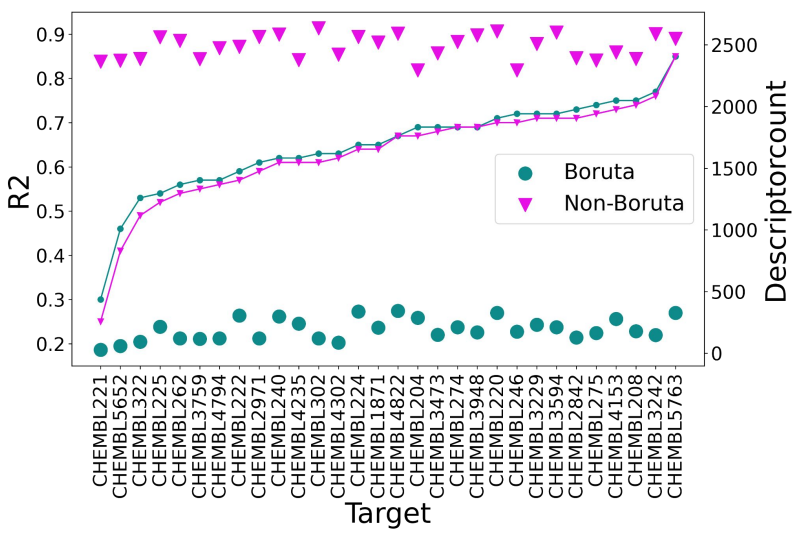
Applying the Boruta algorithm to the large set of descriptors and filtering out the best features enables modelers to build models with fewer descriptors, which saves computational costs and decreases the overfitting issues (refer image to see our analysis on compound sets: ADMET-related targets sourced from the Therapeutics Data Commons along with 30 randomly chosen ChEMBL benchmark datasets with large number of descriptors versus the ones selected by Trainer Engine after applying the needed constraints and Boruta algorithm etc.).
We assessed the performance of Boruta feature selection and hyperparameter selection in the study, “Building machine learning models using relevant features” which was later presented as a poster. These high-quality models perform better with relevant feature set and pre-selected hyperparameter (mTryRatio, maxnodes) definitions which can be further optimized and fine-tuned. Please visit the poster's link for more details on hyperparameter selection.
Choosing the best performing method
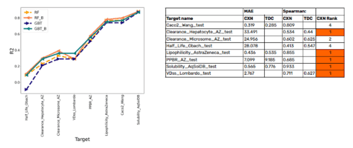
Within Trainer Engine, the Analyze page has a flexible and configurable view that supports visualization, comparison, and assessment of model details and accuracy measures. This allows modelers to narrow down on which methods (Random Forest, Gradient boosted trees etc.) work best with a given dataset, and guides users on how to best finetune the hyperparameters in combination with those methods. Once the best performing model is identified, it can easily be deployed to production.
Integration with Design Hub
Trainer Engine’s integration into Design Hub is an excellent example of this functionality. Virtual compounds in Design Hub can access property calculations generated by ML models in Trainer Engine in the form of a plugin, and exposure to select models can be controlled from within Trainer Engine through deployment. This ensures that users are accessing the most accurate and robust model for any given property.
Future improvements
To further support the ML research needs in drug discovery, Chemaxon plans to further enhance Trainer Engine with deep-learning frameworks like DeepChem. Our goal is to continue the support of the end-to-end needs of ML users from beginners to experts with the latest technology stack, user-friendly interface and make small molecule space computable.
[1] Kursa MB, Jankowski A, Rudnicki WR, Boruta—a system for feature selection. Fundam. Inform. 2010; 101(4):271–285. doi: 10.3233/FI-2010-288