
 Jan-C. Christopherson
Jan-C. Christopherson
Billion scale libraries are here - to stay
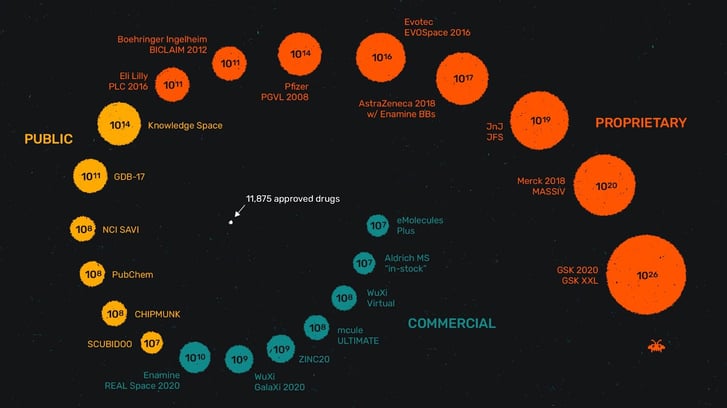
Figure 1. The chemical space of available small molecules is continuously increasing.
In recent years the exploration of chemical space has advanced to a new scale, with libraries often spanning into the billions of molecules. This is spurred by technological advancements on all sides, including cloud computing availability, novel screening methodologies like DEL and automated laboratories.
The pervasiveness of “synthesis on demand” libraries makes it feasible to expand these investigations beyond in silico methods.
As AIML approaches gain traction, it seems inevitable that interest in libraries of this scale will continue to grow to act as a source of input data.
Unique challenges, and intriguing new representation possibilities
This scale of library presents a new set of challenges to the informatics infrastructure used to interrogate them. Most RDBMS cartridges and other similar tools would be hard pressed to store and search on this scale in a way that complies with both the speed expectation of the user as well as their hardware limitations.
A number of non-enumerated formats have been developed that compress the volume of data needed to represent the library. A common method to do this is to represent the library as a set of starting materials, combined with a chemical transformation such as a reaction. This allows for a small set of starting materials to represent a much larger library.
Storing libraries this way requires new search methods be developed in order to search both the starting materials as well as their combination into a product. These methods include LEAP2 and FTrees, which we’ve previously discussed in greater detail.
In order to get to this stage we have to first create such a library and process it; there are two common methods of doing so.
The first is to replace reacting atoms with labels to indicate where the transformation will occur, and the second is to store the vanilla reactants and use a chemical transformation engine to perform structure based matching and transformation as is needed.
Interrogate the product space efficiently with Chemical Space Docking
Docking is a common tool in most drug discovery teams’ arsenals that should not require much introduction to the reader. The ability to determine potential poses of a small molecule in the context of the target protein of interest, and determine a relative preference between a number of potential candidates through the generation of docking scores is a common step in prioritizing ideated compounds.
While it is relatively low in computational effort compared to physics based modeling such as FEP or other quantum mechanical methods, it remains challenging to perform at the scales of these new libraries.
Recently, an approach named “Chemical Space Docking” has been gaining interest as an efficient means to interrogate such large libraries using docking methods. A full introduction to the approach is beyond this piece, but I would like to encourage the reader to take a look at some of the references if they require a primer.
In brief, the method uses an iterative sequence of fragment based docking procedures. At each step, the best results from the previous iteration are selected, and then enumerated with other fragments known to be compatible with their reactive terminal end. The previous iteration’s best poses are used as constraints, making the next docking iteration more computationally efficient by reducing the number of poses to be sampled.
Diagram the process
.png?width=800&height=1338&name=Wiley%20Illustration%20(1).png)
Diagram 1. Chemical space docking
Creating an appropriate representation is a cumbersome prerequisite
Implementing such a workflow is no doubt a difficult, yet worthwhile task. Given a set of possible reactants, how can we easily identify and keep track of those that can undergo the desired transformation?
A number of commercially available libraries may be available in such representations, however a chemist might want to look in their organization’s proprietary compounds in such a search. Additionally, using only such public information does not grant an organization an edge over competitors in their therapeutic target space.
Having the ability to perform an appropriate set of find-and-replace operations in a chemical context is then necessary. Chemaxon’s Reactor is a tool specifically designed for this purpose. It allows the user to input the desired matching conditions and modified output representation with best-in-class chemical accuracy. This allows you to reliably create input data for chemical docking model workflows.
Case study: Amide synthesis from carboxylic acid
A small case study was prepared, performing amide synthesis from a carboxylic acid.
We filtered the first 1000 small molecules from ChEMBL, having first removed all empty structures.
Diagram 2. provides a step-by-step walkthrough of the followed procedure. As well as the additional effort in configuring multiple steps in the process, a few measurable quantities stood out.
First, the final step in the R-group capping method was several orders of magnitude slower than the pre-filtered structure recognition based method.

Diagram 2. Amide synthesis from carboxylic acid
Second, the enumeration using R group or similar end caps results in the generation of far more products. While this might seem appealing at first, we actually believe that it will simply lead to far more unreasonable compounds that either a user or a different filtering process will later have to remove.
Inherent limitations of Synthon representations
One method of representing reactive sites is to remove the atoms that will be removed by the transformation, and replace them with an “R” group representation. These may be referred to as “Synthons”.
While this provides a straightforward way to keep track of reactivity for that particular reaction, there are a number of drawbacks:
- It raises questions about how representations should be handled in case there are multiple matching moieties on the reactant
- Out of context, it removes what could be important information about what the reacting site is
- If updates are made to the reactivity rules, it could be difficult to backtrack and regenerate the matching set of reactants
- If multiple reactions are in scope, it raises questions about how to treat a compound that can undergo multiple different reactions
- If multiple R groups are attached to the same compound, this requires the management of an R group library
- Alternatively, storing the same reactant multiple times with different R groups reduces storage efficiency and could lead to redundant docking computation if not carefully managed
Simple approaches will often apply an R-group to any matching moiety, however as chemists we know that that in itself does not guarantee reactivity. Applications that can calculate the properties of either the entire structure or a substructure to be used as filtering or prioritization conditions will improve the accuracy of the reactivity prediction.
While many commercially available libraries have undergone validation through many years of knowledge gathering (often reported along with library-wide percentage synthetic success rates), most organizations trying to create their own libraries do not have the resources to perform such exhaustive validation.
It’s then important that the software tools that perform the matching assist the user in predicting the likelihood of success, from simple approaches that use molecule properties as candidate criteria to more specific, often machine-learning supplemented retrosynthetic tools.
Reaction based combinatorial representations are useful beyond Chemical Space Docking
As new approaches continue to be developed, we see more use cases for the representations that we have been discussing.
One of the current most prominent use cases is that of DNA encoded libraries. The libraries are intrinsically combinatorial, and as such all of the tools to generate their representations and perform successful searches against these libraries that we have discussed are also relevant to them.
Tackling the challenge of large enumerations
At Chemaxon, we are interested in continuing our investigation of the benefits and challenges of large scale enumerations.
While the computational resources needed to perform combinatorial enumeration may at first appear challenging, the availability of cost-effective platforms such as AWS Spot Instances means this is not insurmountable. This leads naturally to questions of appropriate batching, and management of the workflows being executed. In some cases, task management and distribution services native to cloud services, such as load balancers, may be preferred. In other cases, tools more tightly coupled to the enumeration service, such as the Task Manager capabilities of JChem Microservices Reactor, can be beneficial to the user.
Was this post interesting?
