 Vincent Zoete
Vincent Zoete
 Marta A. S. Perez
Marta A. S. Perez
 Antoine Daina
Antoine Daina
SwissDrugDesign – Aim, history and content
The SwissDrugDesign project was initiated in 2011 by the Molecular Modeling group at the Swiss Institute of Bioinformatics. Led by Prof. Olivier Michielin and Prof. Vincent Zoete, it offers free, cutting-edge online tools for computer-aided drug design (CADD) to support researchers, students and educators in their work.

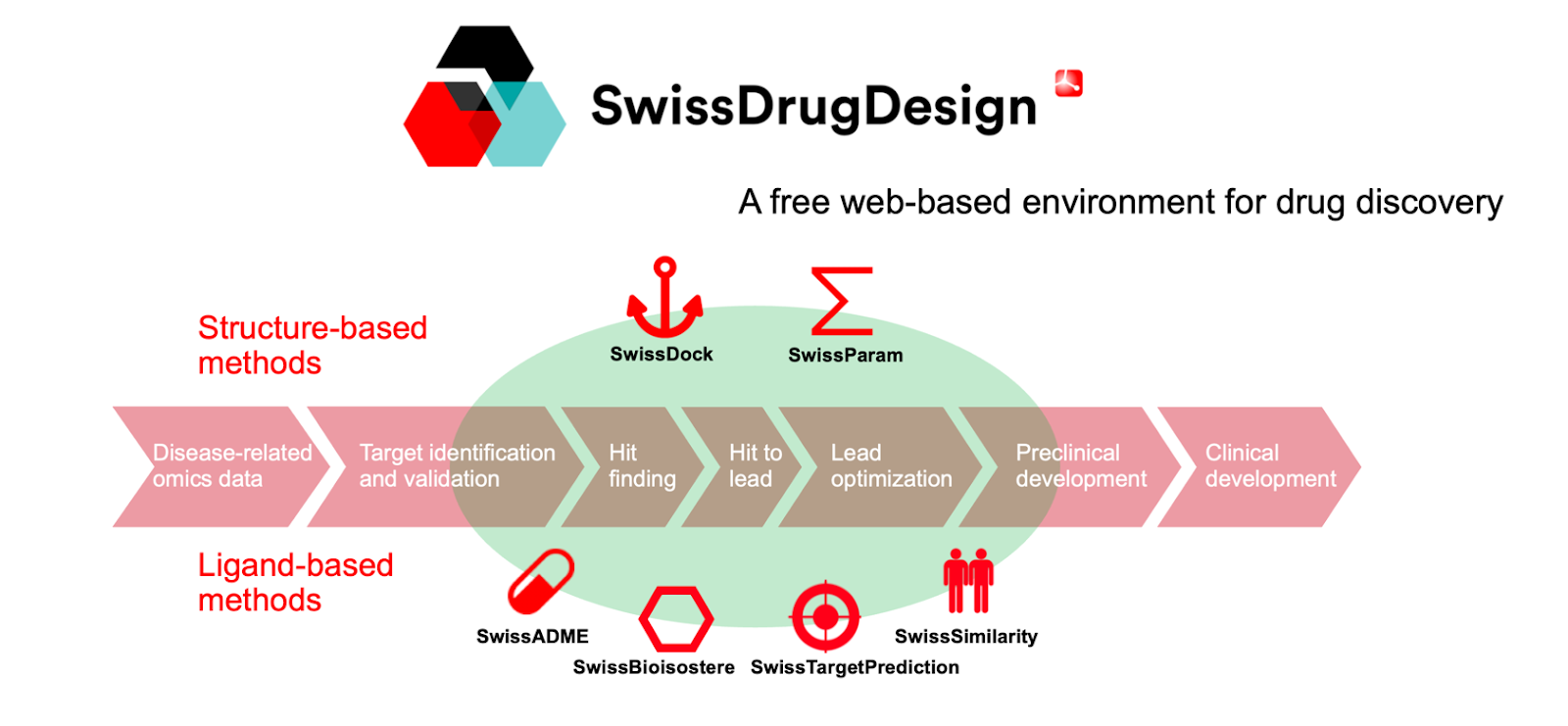
Fig 1. Drug discovery workflow. The SwissDrugDesign environment provides useful tools addressing several steps, notably hit finding, hit to lead and lead optimization. Created by Alessandro Cuozzo
The project was launched in 2011 with SwissDock, a web platform for predicting binding modes of drug-like molecules on proteins. Initially powered by the in-house EADock Dihedral Space Sampling (DSS) docking engine, SwissDock underwent a significant revamp in 2024. It now integrates Attracting Cavities, another in-house engine, alongside the renowned AutoDock Vina docking program. Users can upload their molecules and protein structures or choose them by browsing public databases like ChEBI or the PDB interactively. Both are automatically prepared for the docking. The web site includes a dynamic 3D interface that lets users select protein regions for docking. Results, including predicted ligand-protein interactions and solvent-accissible surface, are visualized interactively on the site or can be downloaded for deeper analysis.
The SwissParam tool, launched in 2011, automates the generation of topology and parameter files for small drug-like molecules, enabling seamless integration with molecular mechanics packages such as CHARMM and GROMACS. Originally developed to meet the specific needs of EADock DSS, SwissParam quickly proved useful for researchers in structure-based drug design employing molecular mechanics methods. In 2023, SwissParam underwent significant updates to broaden its functionality. Key enhancements include the ability to draw molecules of interest directly online. It also introduced features to automatically evaluate multiple microspecies, accommodating pH-dependent molecular behavior. Additionally, SwissParam now supports several parameterization approaches, offering users greater flexibility in their molecular simulations.
These two structure-based CADD tools were joined in 2013 by SwissBioisostere, the first ligand-based approach in the SwissDrugDesign suite. SwissBioisostere is a comprehensive database of unique molecular replacements, enriched with detailed bioactivity and physicochemical properties, together with chemical and biological contexts derived from literature and other resources curated and recorded in the ChEMBL database. This tool primarily supports hit-finding and lead optimization but can also be applied to other medicinal chemistry tasks. SwissBioisostere features an intuitive web-based interface that dynamically displays possible molecular fragment replacements based on user queries, along with some physico-chemical properties, their occurrences and link to the related medicinal chemistry literature. In 2021, SwissBioisostere received a major update, expanding its collection to include 25 million molecular replacements, further consolidating its utility for the exploration of chemical diversity and molecular design in drug discovery.
Ligand-based CADD relies heavily on the similarity principle, which holds that two similar compounds are prone to exhibit similar bioactivities. Building on this principle, the SwissSimilarity web platform was launched in 2016, offering a user-friendly tool for ligand-based virtual screening. It allows users to identify compounds similar to a query molecule, potentially sharing activity on the same protein targets. SwissSimilarity employs diverse molecular similarity definitions, leveraging chemical structure-based and shape-based fingerprints, scaffold-based comparisons, and pharmacophore matching. This versatility ensures robust screening of chemical libraries for hit identification and lead optimization. SwissSimilarity was updated in 2022 to enhance its interface usability, introduce new molecular fingerprints, and expand its screenable chemical collections to include tens of millions of compounds. These span drugs, bioactive molecules, commercially available compounds, and tangible molecules, significantly broadening the tool's applicability in drug discovery and medicinal chemistry.
The similarity principle can also be used to perform reverse screening, by predicting the possible protein targets of bioactive small molecules. This approach is useful for identifying not only the primary targets of drug-like compounds but also their secondary targets, providing insights into potential toxicity and unwanted effects, but also opportunities for drug repurposing. SwissTargetPrediction, launched in 2014 and updated in 2019, was designed with this purpose in mind. This machine-learning-empowered reverse screening tool is able to predict protein targets by comparing (in 2D and in 3D) any query compound submitted by the user to a curated library of approximately half a million compounds with known bioactivities, sourced from the ChEMBL database.
All the aforementioned tools primarily address how small molecules interact with protein targets. However for drug discovery, it is also essential to consider the pharmacokinetic aspects, i.e. how the body processes drugs, focusing on their absorption, distribution, metabolism, and excretion. Addressing this need, SwissADME, launched in 2017, offers free access to a suite of fast yet reliable predictive models for evaluating physicochemical properties, pharmacokinetic behaviors, drug-likeness, and medicinal chemistry friendliness. These include in-house methods such as the BOILED-Egg model for predicting passive absorption and brain penetration, iLOGP for partition coefficient calculations, and the Bioavailability Radar for quick visual assessments of druglikeness.

Fig 2. The SwissDrugDesign environment for CADD. Aim of each single tool. Arrows represent interoperability between the tools; the output of a tool can be used as input for another tool with “one-click” on the corresponding icon. Adapted from the International Journal of Molecular Sciences.
Chemaxon and SwissDrugDesign

Fig 3. Implementation of SwissDrugDesign. Example of the SwissSimilarity website for ligand-based virtual screening. A WebPy web service makes the frontend communicate with the backend code, which performs the necessary calculations. Some steps require JChem engine functions that are called the JChem microservices. Besides, an instance of MarvinJS embedded on the website allows the user to sketch any molecular structure and its web services performs the translation into SMILES, which is the real input of the virtual screening process.
Successfully managing such a high volume of tasks for the scientific community demands meeting several critical requirements. First and foremost, processing millions of molecules from diverse sources — ranging from large databases to individual user submissions — necessitates robust and efficient cheminformatics pipelines. These pipelines must systematically and rapidly curate the input data, transforming it into formats suitable for computational analysis. Additionally, they must ensure the repeatability of processes and consistency of the results, delivering reliable and reproducible outcomes at scale. The SwissDrugDesign projects achieve these objectives by leveraging standard and renown cheminformatics suites such as RDKit, CDK, and Open Babel, alongside with Chemaxon tools for several critical tasks. One key application of the latter is the curation of molecular data to produce standardized, thus reproducible and comparable, canonical SMILES. For SwissDrugDesign, this process involves, but is not limited to, removing extraneous ions and accessory molecular entities to retain a single main molecule per entry, generating its predominant microspecies in a neutral state and at a specified pH, and producing a canonical SMILES in Kekulé form. Notably, the specialized functionality of SwissBioisostere, which suggests potential replacements for molecular fragments, necessitates precise management of information about the connections — specifically, the bonds — linking these fragments to the rest of the molecule. This requirement is efficiently addressed using Chemaxon's extended SMILES format, known as CXSMILES, together with the smart R-group capacity of Marvin, which provides the capability to handle such detailed connection data easily. Additionally, if required, multiple 3D conformations of the molecule are generated and stored in MOL2 format. Importantly, all of these steps must be applied consistently to both the molecules used to generate models and perform calculations (for example in direct or reverse screening datasets) as well as to the users' own molecules. This ensures uniformity and reliability across the entire process, maintaining high standards of data integrity and reproducibility. Chemaxon’s JChem and Marvin tools are integral parts of these workflows, as they comprehensively address all the necessary steps, enabling seamless and coherent execution with consistent coding practices. This ensures a streamlined and reliable data curation process, which is vital for reproducible and high-quality outputs.
Beyond the calculation aspects, the website’s user experience (UX) is also a crucial factor to consider. Key aspects include a well-designed, friendly graphical interface, simplicity and clarity paired with comprehensive input and output pages. In the context of CADD, user-friendliness extends to enabling the sketching of molecules for input, seamlessly converting them into the required formats for calculation engines, generating high-quality images for both input and output molecules, and ensuring rapid response times. This responsiveness is essential for delivering a smooth and efficient UX. In addition, the underlying technology supporting these objectives must be both robust and user-friendly, ensuring optimal website availability while minimizing the time and effort required for maintenance. In the SwissDrugDesign tools, this objective is achieved through the use of Chemaxon’s MarvinJS sketcher and its dedicated MJS web service for drawing molecules and converting them into the correct format. Additionally, Chemaxon’s JChem suite of web microservices handles various other tasks, such as generating curated canonical SMILES, managing pH effects, generating reliable 3D conformations and creating images for both input and output molecules. The MarvinJS sketcher, being JavaScript-based, can be seamlessly integrated into web pages, providing a smooth coding experience. Crucially, the adoption of web microservices to manage the cheminformatics tasks surrounding model execution ensures the tool's efficiency and speed. This approach eliminates the need for time-consuming launches of local programs for each molecule and each run. Moreover, web services offer the advantage of easy sharing across different client web tools, relieving the technical burden of installing and maintaining local programs of possibly different versions for each individual web site. This streamlined, scalable solution enhances both accessibility, performance, at a reasonable maintenance cost.
The SwissDrugDesign web tools can be found at the following URLs: Swissdock.ch, SwissParam.ch, SwissBioisostere.ch, SwissSimilarity.ch, SwissTargetPrediction.ch and SwissADME.ch. They are completely free to use, require no login, and deliver results under the CC BY 4.0 license. Each year, they handle approximately 5 million jobs, for a diverse community of over half a million unique users.
Was this article useful?

Vincent Zoete obtained an engineering degree in chemistry from the ENSCL (Lille, France) in 1995, alongside a DEA in Organic and Macromolecular Chemistry. He started his PhD in Organic Chemistry that same year at the Laboratoire de Chimie Organique et Macromoléculaire, University of Lille I. Concurrently, he obtained a Master’s degree in Drug Design in 1996 in Lille. After a one-year hiatus in 1997-1998, during which he served as a Navy Officer at the French Navy Headquarters in Paris, he completed his PhD in 1999. Following this, he moved to Strasbourg to undertake a postdoctoral fellowship in Molecular Modeling within Martin Karplus's group. During this time, he also collaborated with Enanta Pharmaceuticals (Cambridge, MA, USA). In 2003-2004, he furthered his postdoctoral research by working jointly with Markus Meuwly’s group in Basel and Martin Karplus's group in Strasbourg. In 2004, he joined the Swiss Institute of Bioinformatics (SIB) as a Research Scientist in the Molecular Modeling group of Olivier Michielin and was promoted to Associate Group Leader in 2011. In 2017, he was appointed Assistant Professor in the Department of Oncology at UNIL-CHUV and the Ludwig Institute for Cancer Research, while also serving as a Group Leader at SIB. In 2022, he achieved the rank of Associate Professor.
Vincent Zoete specializes in developing innovative methods in protein engineering and computer-aided drug design, applying them to internal research projects and collaborations with academic and industrial partners. Over the course of his career, he has co-authored approximately 170 research articles and reviews, along with seven book chapters.

Dr. Marta A. S. Perez is a Senior Research Scientist at the Swiss Institute of Bioinformatics (SIB), where she brings innovations in biomedical research, with a focus on computational drug design and immuno-oncology. She holds a PhD in Chemistry from the University of Porto (2013), followed by postdoctoral research at both École Polytechnique Fédérale de Lausanne (EPFL) and Ludwig Cancer Research. Since joining SIB in 2019, Dr. Perez has applied advanced data-analytical techniques, molecular modelling, and computational simulations to advance projects in drug discovery (SwissDrugDesign) and immuno-oncology (including collaborative translational research with Lausanne University Hospital, CHUV). Dr. Perez has co-organized international scientific conferences and is regularly presenting her work to diverse audiences. Her academic contributions include over 30 publications in high-impact journals, more than 1000 citations, and two patented innovations. Additionally, she acts as a peer reviewer for several prestigious scientific journals.

Antoine Daina holds a Federal Diploma of Pharmacist and a PhD in Pharmaceutical Sciences from the University of Geneva. He spent three years as a computational chemist at Syngenta Crop Protection, in the Chemistry Research Department, and three years at the School of Pharmaceutical Sciences in Geneva as Senior Scientist and Lecturer. Thern in 2012 Antoine joined the Molecular Modeling Group at SIB, Swiss Institute of Bioinformatics, where he is now in charge of developing and providing novel methods for computer-aided drug design, which are applied to support academic and industrial drug discovery. His main researches focus on chemoinformatics and machine-learning models for physicochemistry, pharmacokinetics, pharmacodynamics, virtual screening and ADME. He also teaches drug design to Master's and Doctoral students and is involved in the development of educational tools for high schools and the general public.